We know that [NiCl4]2- can potentially take on two different geometries: a tetrahedral and/or square planar.
Following the procedure established for the tetrahedral [NiCl4]2- determine the energy of the square planar [NiCl4]2- (this will have D4h symmetry)
You will need to:
compute square planar [NiCl4]2- at the 3-21G level, on the local computer, don't forget that the square planar structure is a singlet!
this optimisation can be a bit problematic because of the D4h symmetry so add into the additional keywords section "opt=z-matrix". When you come to save the job make sure the "save as cartesian coordinates" check-box is NOT selected, do this for the higher level calculation as well.
use the 3-21G optimised geometry to start a 6-31G(d) structure with the extra keywords activated (you will have three extra keywords now, "opt=z-matrix int=ultrafine scf=(conver=9)", don't forget to resymmetrise the molecule! Carry out this calculation on the SCAN server.
carry out a frequency analysis to confirm you have a minima and a population analysis for the MOs and NBO charges. When you set up this job make sure you remove the "opt=z-matrix" extra keyword (as you are not doing an optimisation!)
then use the 6-31G(d) optimised geometry to start a 6-311G(d,p) structure with the extra keywords activated. Carry out this calculation on the SCAN server.
carry out a frequency analysis to confirm you have a minima and a population analysis for the MOs and NBO charges.
Normally structures are optimised in cartesian coordinates, however in some special cases optimising the structures in terms of bond distances and angles is a better method and the "opt=z-matrix" switches this second option on
With the options outlined above these jobs should take less than 10min each, once your job is running if yours takes more than 10min (in the Wall Time column), do NOT wait for it to finish as you will block the queue for others, DELETE the job from the queue and go back and check that you have implemented the above options correctly. If you still have problems ask a demonstrator, don't repeat jobs without understanding what the problem is.
Your 6-31G(d) job should converge fine, however when you do the frequency analysis you should find that you get one negative frequency! This is a good example of how a converged structure can "appear" to be correct but is actually a transition state. Your negative frequency should be about -31 cm-1. A small negative frequency like this is an indicator that your basis set is not quite good enough and that you need to improve it, or it could be a real transition state ... you won't know until you do the higher level calculation. As this is only a small frequency you can use the output of your 6-31G(d) optimisation to start the 6-311G(d,p) job.
Animate the negative frequency ... can you see how two Cl go up and two Cl go down, this mode is turning the square planar structure into a tetrahedral structure!
When the 6-31G(d) and 6-311G(d,p) jobs have completed successfully record the unique DOI for submission as part of the lab.
Now we will consider the energy of these two complexes, which has the lower energy, the tetrahedral (triplet) or the square planar (singlet)? Normally singlet states are lower in energy than triplet states. However, a tetrahedral geometry is normally lower in energy than a square planar geometry.
Record the energy of your calculations on [NiCl4]2-, mine are detailed below. Yours should be very similar, the same up to 6-7th decimal place, if they are not speak to a demonstrator to find out where you have gone wrong!
3-21G
tetrahedral: -3333.34247396 au
square planar: -3333.24307438 au
6-31G(d)
tetrahedral: -3349.14928357 au
square planar: -3349.09694596 au
6-311G(d,p)
tetrahedral: -3349.41619595
square planar: -3349.37765320
The results of a single calculation a reported in atomic units (hartree) so that others can reproduce your work, this is the ONLY reason for reporting total energies. Never compare calculations run using different basis sets, the results are meaningless.
The important results are always energy differences, these must always be evaluated for calculations carried out with exactly the same method and basis set (and ideally exactly the same "additional keywords" when they relate to the evaluation of the energy of the system.)
When reporting the difference in energy between two isomers we always treat the lowest energy conformer as the reference and report the energy of the other conformers as positive energies, ie being higher in energy than the reference. In my case the tetrahedral conformer is lowest in energy and so it is treated as the reference (ie 0.00 kJ/mol).
Energy differences are reported in kJ/mol, or kcal/mol if you are in the USA. Never in atomic units. Thus convert your ΔE into kJ/mol, here is a link to a good web-based converter
It is standard to report qualitative energy differences to 1 kJ/mol and very accurate energy differences up to 2dp (two decimal places) in kJ/mol, ie 0.01 kJ/mol. Thus, we need to know the energies in au to an accuracy that will allow us to report energy differences to the correct level. The important question is how much is 0.01 kJ/mol in au? 0.01 kJ/mol is 0.0000038 au! So when reporting energies in au you must record them up to at least 6dp (recording up to 8dp is better and then drop the last two when reporting the data)
Thus, the square planar structure lies above the tetrahedral structure by
ΔE=261 kJ/mol at the B3LYP/3-21G level
ΔE=137 kJ/mol at the B3LYP/6-31G(d) level*
ΔE=101 kJ/mol at the B3LYP/6-311G(d,p) level
*structure is a transition state
You can also very clearly see the importance of using a reasonable basis set in order to obtain good energies (actually energy differences). However, the geometry is not as sensitive to the basis set and so we use the low level calculations which are much cheaper to give us a good starting geometry.
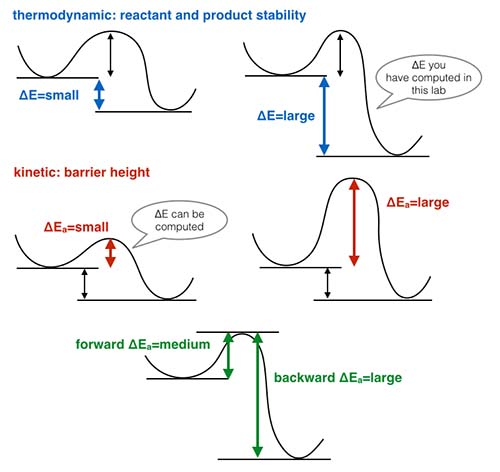
All the calculations carried out here predict that the tetrahedral geometry is the most stable one. Energy differences of 100 kJ/mol are large, thus we would expect the tetrahedral geometry to be almost exclusively the dominant conformer in solution at room temperature. If the energy difference between the conformers is smaller we could expect a small proportion of other conformers to be present in solution, this proportion increases as the energy difference between the conformers decreases.
However, this assumes that the barrier to interconversion between the isomers is small, if this barrier is large then interconversion is unlikely to occur. Typically if an energy barrier is less than 80 kJ/mol it is surmountable at room temperature and multiple conformers could exist.
The image below represents a number of different scenario's related to the relative stability of the complexes and barrier heights for interconversion.
Interpreting Information from the Calculations: Looking at the MOs
Study the MOs from your B3LYP/6-311G(d,p) job, assume the z-axis is out of the plane of the molecule and that the x and y axes lie along the bonds. (this is determined by the symmetry of the molecule and will be explained in more detail next year)
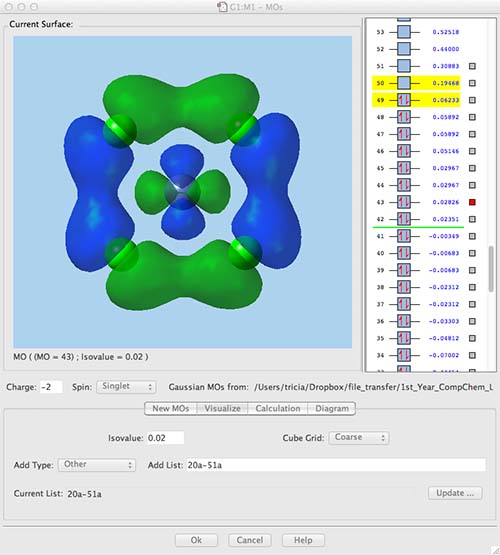
From crystal field theory we would expect a dAO ordering as a LUMO of dx2-y2 character, the dz2 or the dxy MOs next (the order of these MOs is dependent on the ligands) and then at the bottom a degenerate pair of the dxz and dyz.
Does our calculation reflect this? Almost! the LUMO (MO 50) is the dx2-y2 (with quite a bit of antibonding character contributed by the ligands). HOMO (MO 49) is the dz2 orbital. MO 47 and 48 are the degenerate pair of dxz and dyz (antibonding with the ligands). However, MOs 44-46 are completely ligand based MO. What has happened to the dxy MO?? MO 43 is the "missing" dxy MO, the energy of this MO has been substantially lowered by ligand-ligand bonding interactions, moving it out of the "crystal field" manifold. Crystal field theory does not consider the ligand orbitals at all and so it is unable to describe effects like these.
Interpreting Information from the Calculations: Orbital Energy Differences
UV-Vis spectra are due to electronic absorptions exciting electrons from occupied to unoccupied MOs. Typically it is assumed that the HOMO to the LUMO transition is the most important one. However, this is not always true as the electronic states have to have the right symmetry to couple with the incident light (you will learn more about this in your 3rd year).
To a first approximation we might want to relate the HOMO-LUMO energy gap to the visible spectrum:
square planar: 0.06233 to 0.19468 au is 355 kJ/mol =336 nm
tetrahedral alpha:0.02114 to 0.28908 au is 703 kJ/mol =170 nm
tetrahedral beta:0.04159 to 0.17699 au is 347 kJ/mol =345nm
Violet visible light occurs at around 380nm (the full range for violet and blue is approximately 380-500nm) while red visible light occurs at around 750 nm (the full range for orange and red is approximately 590-750nm). If you are interested have a look at the wikipedia entry for the visible_spectrum. Another description can be found here.
One way to approximately estimate the colour of a complex is to use a 19th Century artists colour wheel. There are three primary colours, yellow, blue and red. The secondary colours are created from pairs of primary colours, blue+red=violet, red+yellow=orange, yellow+blue=green. Thus, if we see yellow then the complementary colour violet (red and blue) must be absorbed, if we see blue then orange (red and yellow) are absorbed and if we see red then green (blue and yellow) are absorbed (and vice-versa!)
The calculation gives us a single absorption value, but in reality in a solution the structure is dynamic, moreover, solvent molecules provide a changing environment. These effects broaden the absorption substantially, especially in the case of electronic spectra, which can be extremely broad. A good ball-park figure is a broadening of around 200nm at the base of a "peak". Thus, think of the computed number as the center of a broad peak which ranges ±100 nm.
The first thing to note is that the tetrahedral beta HOMO-LUMO gap is 170nm which even with a broadening to 270nm is well into the ultraviolet range, ie outside of the visible range, and thus will not influence the colour of the complex.
We know that [NiCl4]2- is yellow, therefore it must absorb in the at the violet (red and blue) ends of the spectrum leaving the central yellow part of the spectrum to transmit. This is reflected in the calculation where we can see that the predominant tetrahedral complex will absorb in the high energy region (peak center at ≈350nm and a width from 250-450nm). Thus there will be absorption in the blue/violet region.
Our square planar [NiCl4]2- complex absorbs at about the same wavelength as the tetrahedral structure, and so we could not use the UV-vis spectrum to differentiate between them.
The Crystal Field Model
Traditionally it has been presented that tetrahedral structure d-d excitations are low energy (absorb in the red/orange) and so these complexes are green/blue, while square planar structure d-d excitations are high energy (absorb in the blue/violet) and so these complexes are orange/yellow. However, this empirically based rationalisation, while providing a first approximation, is just not robust enough.
Here we have completely ignored other potential excitations from deeper energy MOs, any relaxation of the orbitals due to a changing electron distribution on excitation, the fact that the formal "dAOs" have large ligand contributions, particularly when the ligands have pi-orbitals to interact strongly with the metal dAOs, and we have ignored charge transfer excitations to or from ligand based orbitals.
"It is well established that these crude crystal field based guidelines cannot be regarded as diagnostic due to pi-bonding and charge transfer which makes a simple crystal-field treatment inappropriate, and unambiguous assignments are difficult"[1] [1] N.N. Greenwood & A. Earnshaw, Nickel, Palladium and Platinum in Chemistry of the Elements, Butterworth Heinemann, Oxford, UK, 1998 pp 1144-1172.
We can account for these other absorptions by carrying out a TD-DFT calculation (TD stands for time dependent), which is beyond your ability at the moment. However, these calculations just confirm that crystal field theory is not sufficient. A robust treatment of the electronic structure (MOs) of the complex must be undertaken before we can understand the absorbances that give rise to the colours of these complexes.
So why use crystal field theory at all? Crystal field theory provides a starting point from which we can build more realistic but also significantly more complex models. A good understanding of crystal field theory is essential before we can start to describe the more complex interactions that occur within a transition metal complex.