
Metal Carbonyls
In Year 1 Inorganic Chemistry, you derived the Molecular Orbital (MO) diagram for carbon monoxide in Structure & Bonding, and in Coordination Chemistry you learned which orbitals in metals and CO are involved in metal-ligand bonding.
Currently in Year 2 in Molecular Orbital theory you covered in lecture 8 ML6 complexes. Also in Year 2 Transition Metal, Coordination and Organometallic Chemistry you are learning more about CO and its modes of binding and stability in complexes, and trends in the physical properties of carbonyl-containing compounds.
In this computational project you will investigate these trends and discuss the underlying chemistry.
First row transition metals, Ti-Fe, are all known to form stable hexacarbonyl complexes of the form [M(CO)6]x±. A nice series of comparable complexes is [Ti(CO)6]2-, [V(CO)6]-, [Cr(CO)6], [Mn(CO)6]+, [Fe(CO)6]2+. These are all isostructural and isoelectronic d6, they are also all low spin because CO is a strong field ligand and all have a multiplicity of 1.
Select three from this series, predict a trend that may be observed or computed, and investigate this computationally. Perhaps collaborate with a friend and each choose different molecules (you should chose one that you both study to ensure you are consistent).
- decide which properties you want to compare, you could consider bond-lengths, vibrational frequencies, MO energies, the octahedral splitting parameter, atomic charges ... Consider at least 2 properties. Document your predictions in your wiki!!
- optimise, and carry out a frequency analysis to confirm you have minima for the three TM-carbonyl complexes you have chosen
- symmetrise to Oh symmetry
- check your charge!
- check the multiplicity, remember multiplicity is (2S+1) where S is the total spin on the molecule, so for these systems which are all spin paired S=0 and multiplicity=1.
- you will need to use a PP on the transition metal and a normal 6-31G(d,p) basis on the C and O atoms. Include the additional input part as shown in the image below, change "M" for your metal
- to save time generating the input structures, optimise one structure first, then you can simply edit the log file and change out the central metal before resubmitting as a new *.com/*.gjf file. Be sure you re-add the additional input part changing the PP!
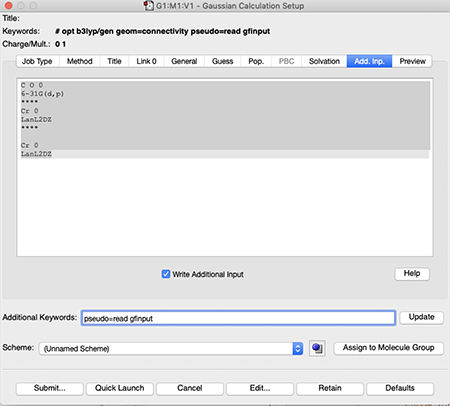
C O 0 6-31G(d,p) **** M 0 LanL2DZ **** M 0 LanL2DZ
- now analyse the properties you have chosen to compare, format your data in an accessible and meaningful way, think about having clear tables and graphs. Comment on reality vs your predictions, were you right or wrong? Justify or explain the trends you observe.
- discuss whether it is possible to analyse the totally symmetric C-O vibrations for these complexes?
- compute the MOs for at least one of your molecules, visualise all the occupied valence MOs and the lowest 5 unoccupied MOs, you do not need to reproduce them all. If you don't know what the valence MOs are ask!
- in your wiki present 3 valence MOs from those you have visualised. Cover a range of MOs showing bonding and antibonding character, do not choose MOs from the same degenerate set.
- draw a LCAO MO diagram for each of these orbitals (in chemdraw). Describe and annotate the interactions occurring in the MOs, identify the overal MO character. Refer to the "read first!" lab web-page and your MO course notes for the format of this description. You might want to revise Lecture 8 from your MO course.
- optional Comment on or analyse or describe some aspect of this system which you found interesting.